SemiBin
If you use this software in a publication please cite:
Pan, S.; Zhu, C.; Zhao, XM.; Coelho, LP. A deep siamese neural network improves metagenome-assembled genomes in microbiome datasets across different environments. Nat Commun 13, 2326 (2022). https://doi.org/10.1038/s41467-022-29843-y
The self-supervised approach and the algorithms used for long-read datasets (as well as their benchmarking) are described in
Pan, S.; Zhao, XM; Coelho, LP. SemiBin2: self-supervised contrastive learning leads to better MAGs for short- and long-read sequencing. Bioinformatics Volume 39, Issue Supplement 1, June 2023, Pages i21–i29; https://doi.org/10.1093/bioinformatics/btad209
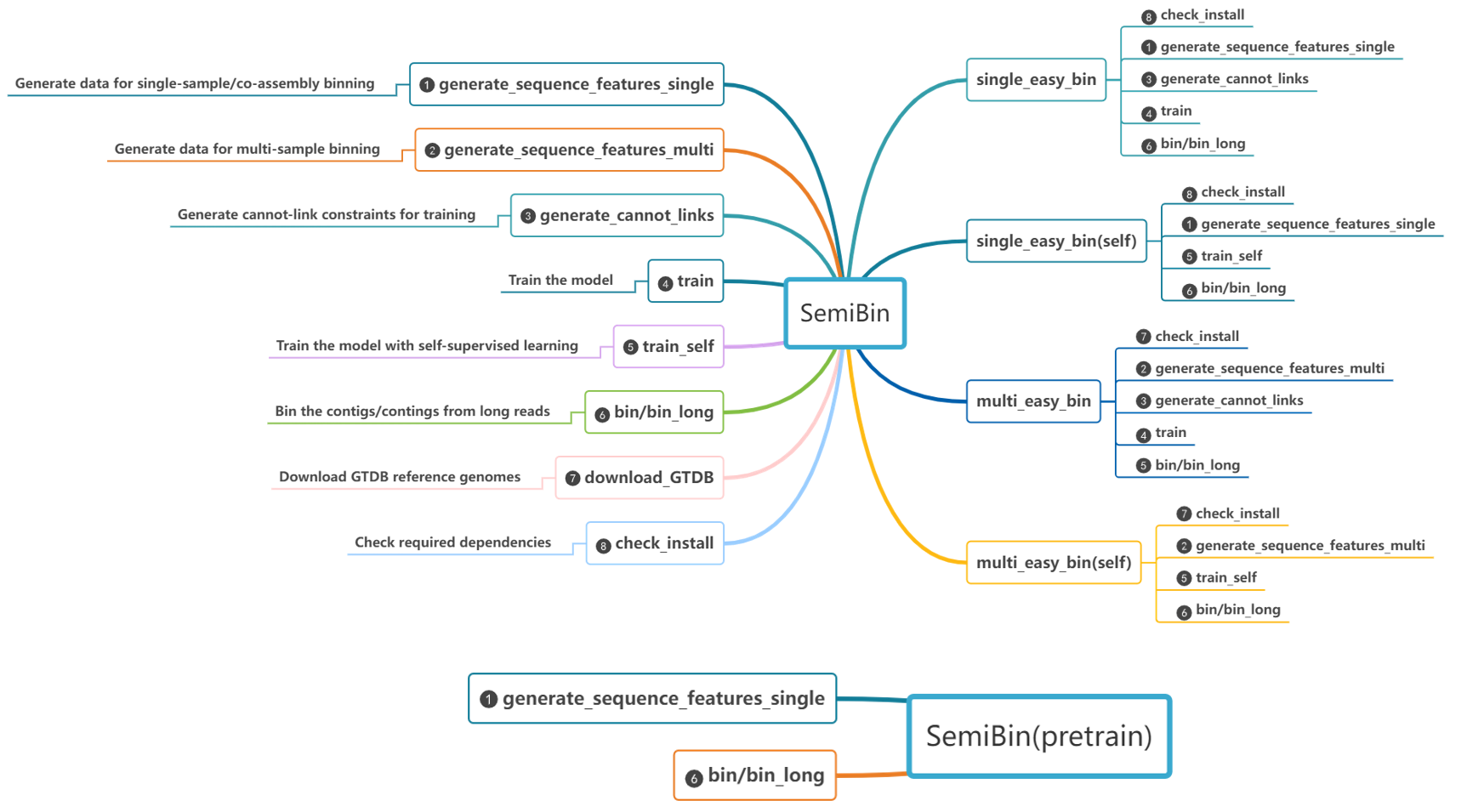
SemiBin is a command line tool for metagenomic binning with semi-supervised siamese neural network using additional information from reference genomes and contigs themselves. It supports single sample, co-assembly, and multi-samples binning modes.
SemiBin2
When you install the SemiBin package, the SemiBin2 command is available.
See SemiBin2 for more details on the differences from the older SemiBin1 interface.
Tutorial
We have a tutorial available with example datasets that runs through most of the functionality of SemiBin:
https://github.com/BigDataBiology/SemiBin_tutorial
Install
The current recommended way to install SemiBin is to use pixi. Pixi will use the packages from conda-forge and bioconda to install SemiBin and its dependencies.
pixi init
pixi add bioconda::semibin
See Install for details and how to install from source or how to enable GPU usage.
SemiBin Examples
See the usage page for a more in-depth overview of how SemiBin can be used.
Single-sample binning
[How to generate inputs to SemiBin]
If your assembled contigs are in a file called S1.fa (contig file in FASTA format) and you mapped reads and sorted the output to generate the BAM file S1.sorted.bam, then you there are two options.
1. Using a pre-trained model. This is the fastest option and should work the best if you have metagenomes from one of our prebuilt habitats (alternatively, you can use the global "habitat" which combines all of them).
SemiBin2 single_easy_bin \
--environment human_gut \
-i S1.fa \
-b S1.sorted.bam \
-o output
2. Learn a new model. Alternatively, you can learn a new model for your data. The main disadvantage is that this approach will take longer:
SemiBin2 single_easy_bin \
-i S1.fa \
-b S1.sorted.bam \
-o output