SemiBin
If you use this software in a publication please cite:
Pan, S., Zhu, C., Zhao, XM. et al. A deep siamese neural network improves metagenome-assembled genomes in microbiome datasets across different environments. Nat Commun 13, 2326 (2022). https://doi.org/10.1038/s41467-022-29843-y
SemiBin is a command line tool for metagenomic binning with semi-supervised siamese neural network using additional information from reference genomes and contigs themselves. It supports single sample, co-assembly, and multi-samples binning modes.
Install
The simplest way to install is to use conda.
conda create -n SemiBin
conda activate SemiBin
conda install -c conda-forge -c bioconda semibin
See Install for how to install from source or how to enable GPU usage.
Single sample binning
Single sample binning means that each sample is assembled and binned independently.
This mode allows for parallel binning of samples and avoid cross-sample chimeras, but it does not use co-abundance information across samples.
Co-assembly binning
Co-assembly binning means samples are co-assembled first (as if the pool of samples were a single sample) and binned later.
This mode can generate better contigs (especially from species that are at a low abundance in any individual sample) and use co-abundance information, but co-assembly can lead to intersample chimeric contigs and binning based on co-assembly does not retain sample specific variation. It is appropriate when the samples are very similar (e.g., a time-series from the same habitat).
Multi-sample binning
With multi-sample binning, multiple samples are assembled and binned individually, but information from multiple samples is used together. This mode can use co-abundance information and retain sample-specific variation at the same time. However, it has increased computational costs.
This mode is implemented by concatenating the contigs assembled from the individual samples together and then mapping reads from each sample to this concatenated database.
Overview of the subcommands
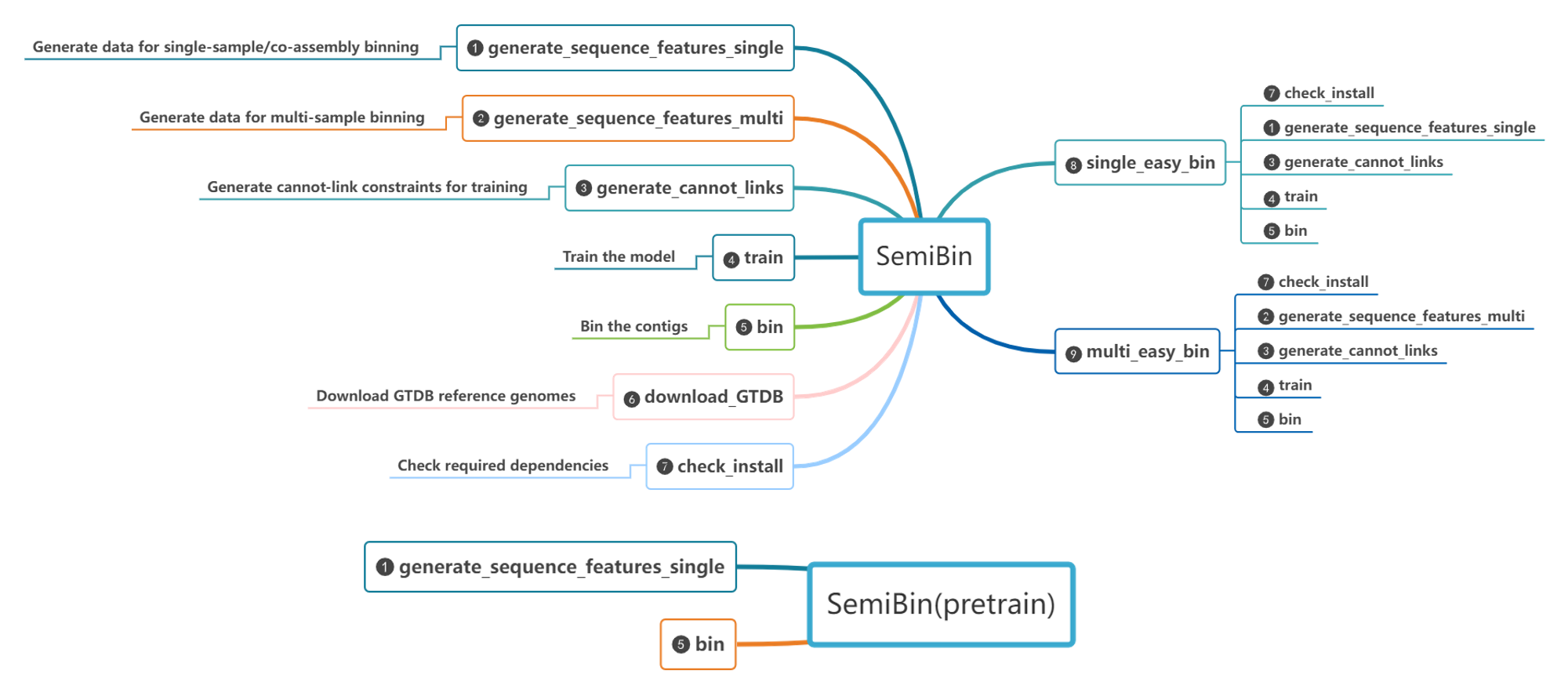
Commands
single_easy_bin
Reconstruct bins with single or co-assembly binning using one line command.
The command single_easy_bin requires the contig file (assembly from reads), bam files (reads mapping to the contig) as inputs and outputs reconstructed bins in the output_recluster_bins directory.
Required arguments
-i/--input-fasta: Path to the input contig fasta file (gzip and bzip2 compression are accepted).-b/--input-bam: Path to the input BAM(.bam) or CRAM(.cram) files . You can pass multiple BAM files, one per sample.-o/--output: Output directory (will be created if non-existent).
Recommended argument
If your data comes from one of the habitats for which we have a prebuilt model, using the --environment argument will use it instead of training a new model.
--environment: Environment for the built-in model (human_gut/dog_gut/ocean/soil/cat_gut/human_oral/mouse_gut/pig_gut/built_environment/wastewater/chicken_caecum/global).
If --environment is not given, a new model is learned (computationally intensive).
Optional arguments to control computational resource usage
-p/--processes/-t/--threads: Number of CPUs used (0, the default, indicates that all CPUs should be used).--engine: device used to train the model (auto/gpu/cpu);auto(default) means that SemiBin with attempt to detect and use GPU and fallback to CPU if no GPU is found.--tmpdir: set temporary directory.-r/--reference-db-data-dir: GTDB reference directory (Default:$HOME/.cache/SemiBin/mmseqs2-GTDB). SemiBin will lazily download GTDB if it is not found there. Note that a lot of disk space is used
Optional arguments to set internal parameters
--random-seed: Random seed to reproduce results.--orf-finder: gene predictor used to estimate the number of bins. Must be eitherprodigal(default sincev0.7) orfraggenescan(which is faster, but cannot be installed in all platforms).
Optional arguments to bypass internal steps
Several internal steps can be bypassed if you wish to compute it outside of SemiBin. For example, calling mmseqs2 for contig annotation takes a lot of time and if you perform it for your dataset independently of SemiBin, you can reuse the results here and avoid recomputaation.
--taxonomy-annotation-table: TAXONOMY_TSV, Pre-computed mmseqs2 format taxonomy TSV file to bypass mmseqs2 GTDB annotation [advanced]. When running with multi-sample binning, please make sure that the order of the taxonomy TSV file and the contig file (used for the combined fasta) is same.--depth-metabat2: depth file generated by metabat2 (only used with single-sample binning).
Optional arguments to set internal parameters (advanced)
Generally speaking, you should use the default values for all these parameters, but they are provided if you want to tune the algorithms. If you find that changing these significantly improves the results of binning, we would appreciate if you got in touch.
--minfasta-kbs: minimum bin size in kilo-basepairs (Default: 200).--recluster: [Deprecated] Does nothing (current default is to perform clustering).--no-recluster: Do not recluster bins.--epoches: Number of epoches used in the training process (Default: 20).--batch-size: Batch size used in the training process (Default: 2048).--max-node: Percentage of contigs that considered to be binned (Default: 1).--max-edges: The maximum number of edges that can be connected to one contig (Default: 200).--ratio: If the ratio of the number of base pairs of contigs between 1000-2500 bp smaller than this value, the minimal length will be set as 1000bp, otherwise 2500bp. If you set -m parameter, you do not need to use this parameter. If you use SemiBin with multi steps and you use this parameter, please use this parameter consistently with all subcommands (Default: 0.05).-m/--min-len: Minimal length for contigs in binning. If you use SemiBin with multi steps and you use this parameter, please use this parameter consistently with all subcommands. (Default: SemiBin chooses 1000bp or 2500bp according the ratio of the number of base pairs of contigs between 1000-2500 bp).--ml-threshold: Length threshold for generating must-link constraints. By default, the threshold is calculated from the contig, and the default minimum value is 4,000 bp.--cannot-name:Name for the cannot-link file (Default:cannot).
multi_easy_bin
Reconstruct bins with multi-samples binning using one line command.
The command multi_easy_bin requires the combined contig file from several samples, bam files (reads mapping to the combined contig) as inputs and outputs the reconstructed bins in the samples/[sample]/output_recluster_bins directory.
Required arguments
-b/--input-bam: Path to the input BAM (.cram) or CRAM (.cram) files. You can pass multiple BAM files, one per sample.--input-fastaand--outputare same as forsingle_easy_bin.
Optional arguments
-s/--separator: Used when multiple samples binning to separate sample name and contig name (Default is:).--reference-db-data-dir,--processes,--minfasta-kbs,--recluster,--epoches,--batch-size,--max-node,--max-edges,--random-seed,--ratio,--min-len,--ml-threshold,--no-recluster,--orf-finder,--engineand--tmpdirare same as forsingle_easy_bin
generate_cannot_links
Run the contig annotations using mmseqs with GTDB and generate cannot-link file used in the semi-supervised deep learning model training.
The subcommand generate_cannot_links requires the contig file as inputs and outputs the cannot-link constraints.
Required arguments
--input-fastaand--outputare same as forsingle_easy_bin.
Optional arguments
-o/--output,--cannot-name,-r/--reference-db-data-dir,--ratio,--min-len,--ml-threshold,--taxonomy-annotation-tableand--tmpdirare same as forsingle_easy_bin.
generate_sequence_features_single
The subcommand generate_sequence_features_single requires the contig file and bam files as inputs and generates training data (data.csv; data_split.csv) for single and co-assembly binning.
Required arguments
-i/--input-fasta,-b/--input-bamand-o/--outputare same as forsingle_easy_bin.
Optional arguments
-p/--processes/-t/--threads,--ratio,--min-len,--ml-threshold,--depth-metabat2and--tmpdirare same as forsingle_easy_bin.
generate_sequence_features_multi
The subcommand generate_sequence_features_multi requires the combined contig file and bam files as inputs and generates training data (data.csv;data_split.csv) for multi-sample binning.
Required arguments
-i/--input-fastaand-o/--outputare the same as forsingle_easy_bin.-b/--input-bamare the same as formulti_easy_bin.
Optional arguments
-p/--processes/-t/--threads,--ratio,--min-len,--ml-thresholdand--tmpdirare the same as forsingle_easy_bin.-s/--separatorare the same as formulti_easy_bin.
train
The train subcommand requires the contig file and outputs (data.csv, data_split.csv and cannot.txt) from the generate_sequence_features_single, generate_sequence_features_multi and generate_cannot_links subcommand as intpus and outputs the trained model.
Required arguments
--data: Path to the input data.csv file.--data_split: Path to the input data_split.csv file.-c/--cannot-link: Path to the input cannot link file generated from other additional biological information, one row for each cannot link constraint. The file format: contig_1,contig_2.--mode: [single/several] Train models from one sample or several samples(train model across several samples can get better pre-trained model for single-sample binning.) In several mode, must input data, data_split, cannot, fasta files for corresponding sample with same order. Note: You can just setseveralwith this option when single-sample binning. Training from several samples with multi-sample binning is not supported.-i/--input-fasta,-o/--outputare the same forsingle_easy_bin
Optional arguments
--epoches,--batch-size,-p/--processes/-t/--threads,--random-seed,--ratio,--min-len,--orf-finderand--engineare the same as forsingle_easy_bin
bin
The bin subcommand requires the contig file and output (data.csv, model.h5) from the generate_sequence_features_single, generate_sequence_features_multi and train subcommand as inputs and output the final bins in the output_recluster_bins directory.
Required arguments
--model: Path to the trained model.--data,-i/--input-fasta,-o/--outputare the same as forsingle_easy_bin.
Optional arguments
--minfasta-kbs,--recluster,--max-node,--max-edges,-p/--processes/-t/--threads,--random-seed,--environment,--ratio,--min-len,--no-recluster,--orf-finder,--engineand--depth-metabat2are the same as forsingle_easy_bin
download_GTDB
Download reference genomes (GTDB).
-r/--reference-db-data-dir: Where to store the GTDB data (default:$HOME/.cache/SemiBin/mmseqs2-GTDB)-f/--force: Whether to download GTDB even if the data is found at the path (default is to not download).
If you download GTDB to a different directory than the default, you should then pass that path to every command (-r) to ensure that it is found.
check_install
Checks whether required dependencies are available (useful for trouble-shooting).
concatenate_fasta
Concatenate fasta files for multi-sample binning.
The contigs are renamed to include the sample name followed by a separator character.
The separator character cannot occur in any of your samples, so if any sample contains the default separator (:), you must change it and pass that information to every command (using the --separator/-s argument).
Required arguments
-i/--input-fasta,-o/--outputare the same as forsingle_easy_bin.
Optional arguments
-s/--separatoris the same as themulti_easy_bin(see comment above).-m: Discard sequences below this length (default:0).